Selected preprints
- J Hay et al.,
"Evaluation of influenza A/H3N2 epidemiology in England during the 2025-26 season",
https://zenodo.org/records/18019825
England has experienced a high growth rate of infections caused by the influenza A/H3N2 K clade.
Antigenic change from the previously dominant clade, a rapid selective sweep evident in genomic data,
and an unusually early start to the season have raised concerns about the potential severity of this year’s influenza season.
We found that
the peak growth rate of influenza infections and the peak time-varying reproduction number has been
largely consistent with previous severe seasons.
Substantial immune escape appears to be unlikely given current epidemiological trends.
In almost all scenarios, an earlier and faster epidemic growth rate leads to earlier depletion of susceptibles
with a dampening effect due to the half term school holiday.
This rapid analysis is intended to support situational awareness.
To support understanding and exploration of model outputs, an interactive visualisation tool was developed and made available online.
- SE Ramos-Onsins and L Ferretti,
"Genetic signatures of adaptation to sudden, extreme, and unprecedented environmental changes",
https://www.biorxiv.org/content/10.64898/2026.02.27.708566
Small natural populations are increasingly exposed to environmental shifts that are unprecedented in speed and intensity on population-genetics timescales. If populations persist, adaptation to these abrupt changes is expected to leave localized footprints of selection around the loci contributing to the adaptive response, in contrast to genome-wide signals produced by demography alone. Here we provide an explicit theoretical description of genetic footprints expected under rapid adaptation from standing genetic variation. We derive exact analytical results for how single-locus sweeps and polygenic shifts driven by sudden environmental changes distort the site-frequency spectrum (SFS) of linked mutations, and we link these distortions to the expected changes in standard diversity measures and neutrality statistics. Our results provide a guideline for detecting and interpreting genetic signatures of adaptation to extreme events, including those associated with climate change, particularly in small populations where adaptation or evolutionary rescue often proceed quickly and from preexisting variation.
- SE Ramos-Onsins, J Ross-Ibarra, M Caceres, L Ferretti,
"Bias in diversity estimators and neutrality tests induced by neutral polymorphic structural variants",
https://www.biorxiv.org/content/10.64898/2026.02.26.708357
Estimators of genetic diversity and neutrality tests derived from the site frequency spectrum (SFS),
are designed to be interpreted relative to a baseline defined by the standard neutral SFS.
In genomic regions strongly linked to a polymorphic structural variant (SV),
deviations from these baselines occur even under strict neutrality, distorting the SFS for linked neutral mutations.
We provide analytical formulae for the expected SFS of single nucleotide polymorphisms conditional on neutral linked polymorphic SVs,
including inversions, deletions, insertions, and introgressions.
We quantify the resulting bias in standard diversity estimators and neutrality tests.
Finally, we discuss approaches to build corrected estimators of diversity and neutrality tests that are unbiased/centered after
accounting for the presence and frequency of the SV.
- F Di Lauro and L Ferretti,
"Preferential attachment and power-law degree distributions in heterogeneous multilayer hypergraphs",
http://arxiv.org/abs/2505.18068
Preferential attachment is the most popular mechanism to explain scale-free degree distributions in growing networs.
However, realistic network models should also include complex connectivity structures
and different types of heterogeneity, such as multiple node types, multiple layers, and hyperlinks with multiple orders (hypergraphs).
Here we study arbitrarily complex network models growing by a linear preferential attachment mechanism.
We show how all these models have a multi-power-law hyperdegree distribution.
For generic connectivity structures, the exponent of the power-law distribution is universal
for all layers and all orders of hyperlinks, and it depends exclusively on the type of node.
- L Ferretti, T Golubchik, F Di Lauro, M Ghafari, J Villabona-Arenas, KE Atkins, C Fraser and M Hall,
"Biased estimates of phylogenetic branch lengths resulting from the discretised Gamma model of site rate heterogeneity",
https://www.biorxiv.org/content/10.1101/2024.08.01.606208v1
For the last 3 decades, the standard model for phylogenetics has included the Gamma4 model of site heterogeneity.
We show that this model has a fundamental issue of bias in branch lengths, and
that the inferred length of a branch increases with the number of tips present in other parts of the tree.
The problem originates from the equally sized rate classes.
We recommend that the use of Gamma4 and other models based on equal rate classes be discontinued.
Unfortunately, data access agreements and approval processes sometimes prevent some of our publications from being preprinted.
Please get in touch if you are interested in ongoing work not listed here.
Selected recent publications
-
A Colubri, D Williams, T Valente, CT Bauch, JM Drake, MC Mills, J Drury, C Fraser, L Ferretti and J Panovska-Griffiths,
“Understanding human behaviour for pandemic preparedness with epigames”,
Nature Health (2026).
For many infectious diseases, transmission occurs when individuals are in close proximity for a sufficient time and
through highly structured social contact networks.
Data on the properties of these networks, including their temporal and spatial structures,
how pathogens spread in them, and how interventions might alter this spread are scarce or
inconsistent and seldom incorporate behavioural features.
We propose a solution that leverages mobile technology to measure contact networks across social settings,
environmental conditions and various contexts, while explicitly integrating behavioural data.
- M Kraemer, JL-H Tsui, SY Chang, S Lytras, MP Khurana, S Vanderslott, S Bajaj,
N Scheidwasser, JL Curran-Sebastian, E Semenova, M Zhang, HJT Unwin, OJ Watson,
C Mills, A Dasgupta, L Ferretti, SV Scarpino, E Koua, O Morgan, H Tegally, U Paquet,
L Moutsianas, C Fraser, NM Ferguson, EJ Topol, DA Duchene, T Stadler, P Kingori,
MJ Parker, F Dominici, N Shadbolt, MA Suchard, O Ratmann, S Flaxman, EC Holmes,
MG Rodriguez, B Scholkopf, CA Donnelly, OG Pybus, S Cauchemez* and S Bhatt*,
“Artificial Intelligence for Modelling Infectious Disease Epidemics”,
Nature 638, 623–635 (2025).
Perspective on the potential applications of AI to infectious diseases.
- M Kendall*, L Ferretti*, C Wymant, D Tsallis, J Petrie, A Di Francia,
F Di Lauro, L Abeler-Dorner, H Manley, J Panovska-Griffiths, A Ledda, X Didelot and
C Fraser, “Drivers of epidemic dynamics in real time from daily digital COVID-19
measurements”, Science 385, eadm8103 (2024).
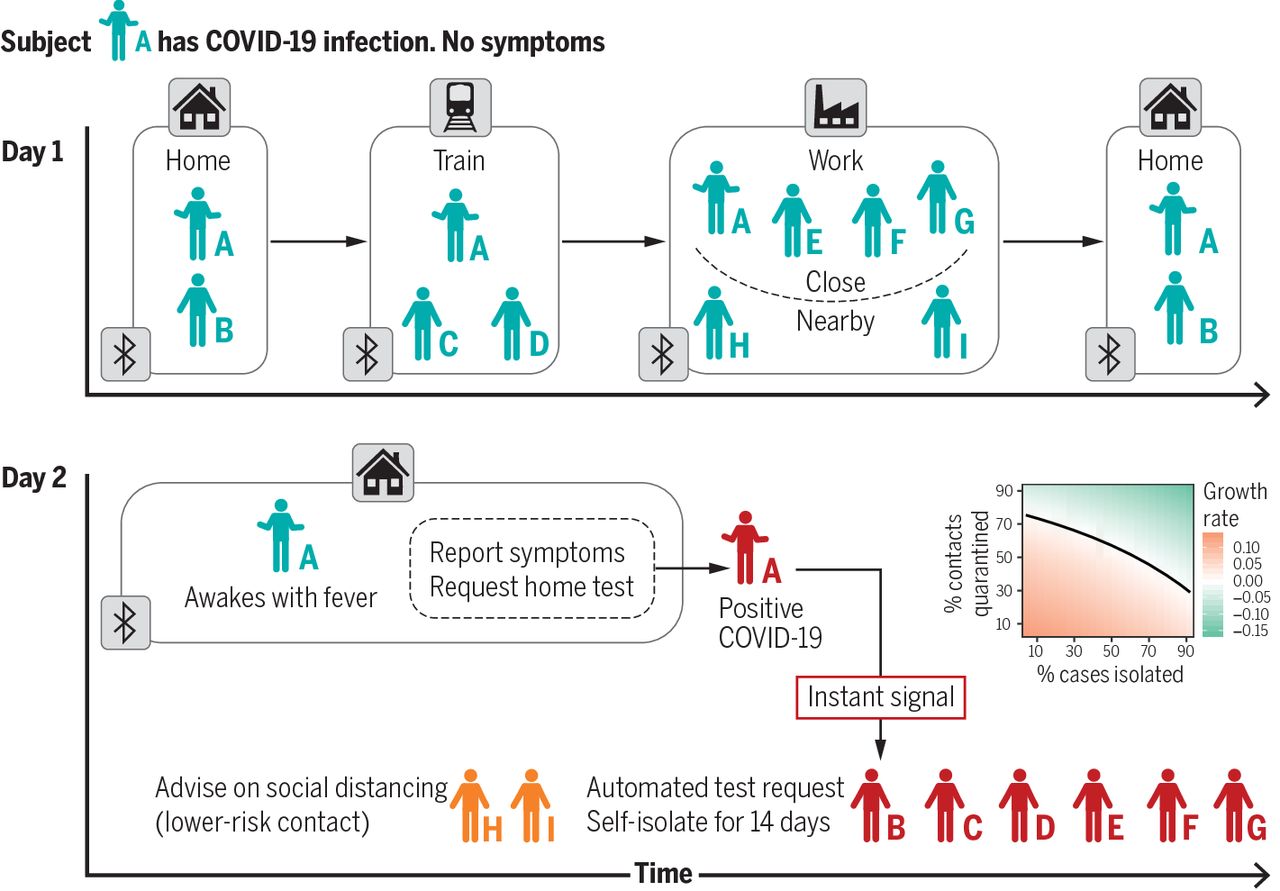
Unprecedented epidemic monitoring of transmissions during Euro 2021 and other events.
Featured on the cover of Science.
- B Singer*, A Di Nardo*, J Hein and L Ferretti, “Comparing phylogeographies to reveal
incompatible geographical histories within genomes”,
Molecular Biology and Evolution 41,
7 msae126 (2024)
First proposal of measures of incompatibility/distance between phylogeographies.
- L Ferretti*, C Wymant*, J Petrie, D Tsallis, M Kendall, A Ledda, F Di Lauro,
A Fowler,
A Di Francia, J Panovska-Griffiths, L Abeler-Dorner, M Charalambides, M Briers and C Fraser,
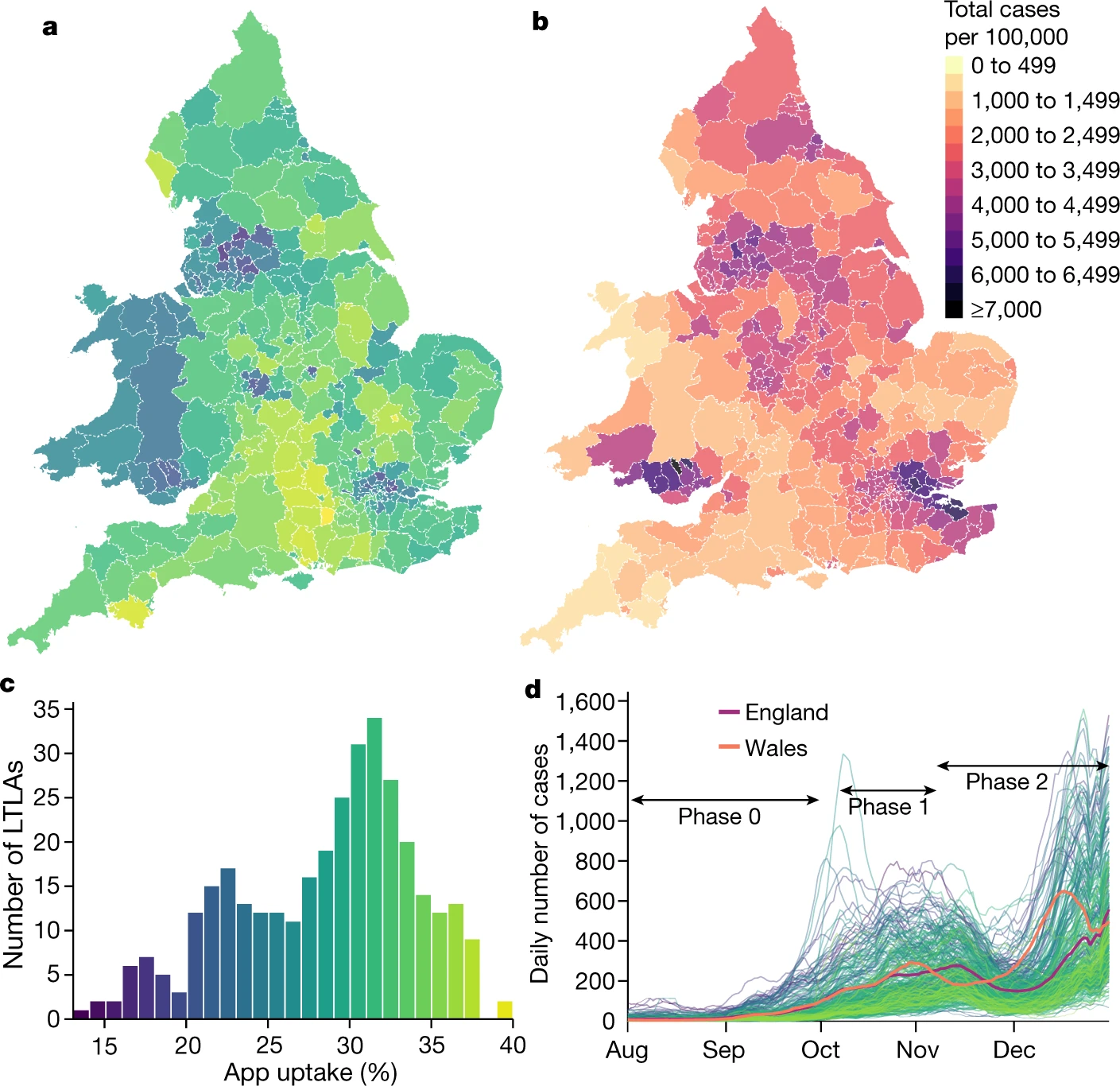
“Digital measurement of SARS-CoV-2 transmission risk from 7 million contacts”,
Nature 626 (7997), 145–150 (2024).
First high-resolution analysis of COVID-19 transmissions vs duration/proximity.
Key paper for precision epidemiology of respiratory pathogens.